NanoporeSignals.jl
NanoporeSignals is a Julia library for working with raw nanopore signals. It currently provides utilities for signal-to-sequence alignments, signal scaling, segmentation and others.
The library is still in active development and should be considered unstable. The functionality related to signal-to-sequence alignment should be relatively more stable and reliable. Some functions are explicitly marked as experimental and should be used especially cautiously.
Installation
julia> using Pkg
julia> Pkg.add("https://codeberg.org/mzdravkov/NanoporeSignals.jl")Example
julia> using XAM
julia> using Slow5
julia> using NanoporeSignals
julia> reads = collect(open(BAM.Reader, "reads.bam"))
julia> alignment = SignalAlignment(reads[1]; pore=:RNA004)
SignalAlignment(read=7da49fe9-041c-4512-8e3c-cd88a7723ec6; seq_range=(15, 432); sig_range=(10618, 15696), reverse=true)
# Get k-mer to signal intervals mapping
julia> kmer_to_sig(alignment) |> collect
126-element Vector{Pair{Tuple{Int64, Int64}}}:
(15, 23) => (15677, 15696)
(16, 24) => (15667, 15676)
(17, 25) => (15660, 15666)
⋮
(422, 430) => (10698, 10715)
(423, 431) => (10623, 10697)
(424, 432) => (10618, 10622)
julia> blow5 = slow5_open("raw_records.blow5", "r")
julia> slow5_idx_load(blow5)
# Get the aligned signal, converted to picoampers.
julia> aligned_signal(alignment, blow5; to_pa=true)
125-element Vector{Tuple{Any, Vector{Float64}}}:
(429, [71.34, 66.81, 72.81, 68.86, 66.37])
(428, [94.74, 94.44, 94.30, 83.48, 89.47 … 95.91, 84.94, 92.98, 94.01, 89.47])
⋮
(22, [63.16, 64.03, 63.60, 61.40, 63.30, 63.16, 64.47])
(21, [70.47, 65.35, 64.47, 65.35, 73.39, 63.60, 64.47, 66.96, 64.33, 76.17])
# Get the aligned signal for all k-mers overlapping the sequence interval [66, 72].
julia> aligned_signal(slice(alignment; from=66, to=72), blow5; to_pa=true)
12-element Vector{Tuple{Int64, Vector{Float64}}}:
(77, [101.76, 101.76, 102.78, 97.37, 101.61 … 100.29, 99.71, 98.39, 94.30, 99.42, 94.59])
(76, [83.48, 76.02, 75.73, 79.24, 76.17, 81.72, 72.51, 73.98, 76.75, 79.53])
⋮
(67, [80.85, 86.11, 83.33, 70.03, 90.35, 88.89])
(66, [101.46, 108.04, 107.60, 112.72, 113.74 … 82.75, 69.4, 77.63, 86.26, 111.26])
# Normalize read signals using their poly(A) levels.
scaled_reads = Dict()
for read in reads
aln = SignalAlignment(read; pore=:RNA004)
raw_record = get_raw_read(read, blow5)
signal = get_pa(raw_record)
polyA = polyA_range(aln, string(BAM.sequence(read)), signal)
if polyA !== missing
from, to = polyA
scaled_reads[BAM.tempname(read)] = signal ./ median(signal[from:to])
end
end
Introduction
Sequence-to-signal alignments
In nanopore sequencing, each current intensity sample point is measuring a k-mer that is passing through the pore at the given moment. The number of neighbouring bases that influence the signal (i.e. the k-mer size) and the representative position (the position within the k-mer that influences the signal the most, a.k.a. k-mer center) depend on the pore model. Due to the irregular speed of the molecule translocation, each k-mer can stay in the pore for different amount of time. Hence, finding the signal corresponding to a given part of the sequence requires the use of complicated signal-to-sequence alignment (a.k.a. resquiggling) algorithms. This functionality is implemented in resquiggling tools such as f5c and Uncalled4.
Both f5c and Uncalled can output the sequence-to-signal alignments in SAM/BAM format, using the si and ss tags. This library allows the parsing of those to get convenient Julia representations of the alignments, based on the IntervalTrees package. The format has been described very nicely in f5c's documentation here.
A DNA signal alignment can look like this: 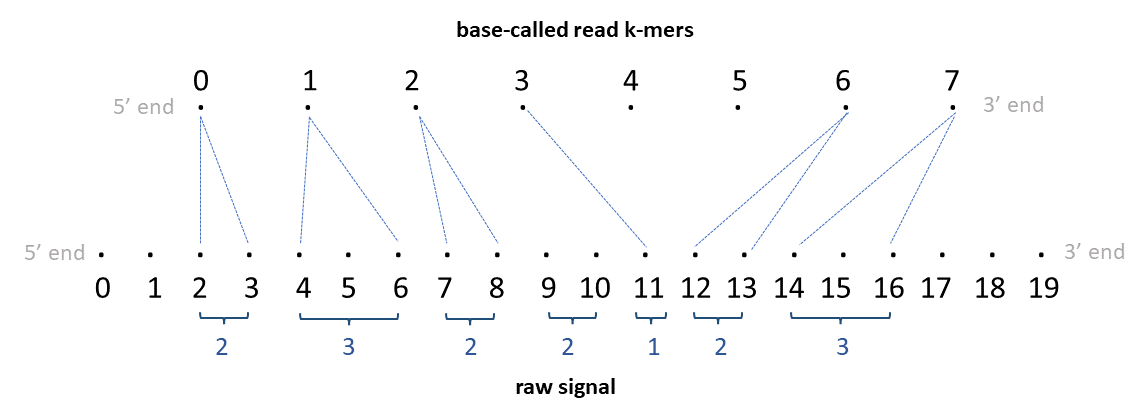
Whereas an alignment in dRNA (where the molecule is sequenced in 3′->5′ direction) will look like this: 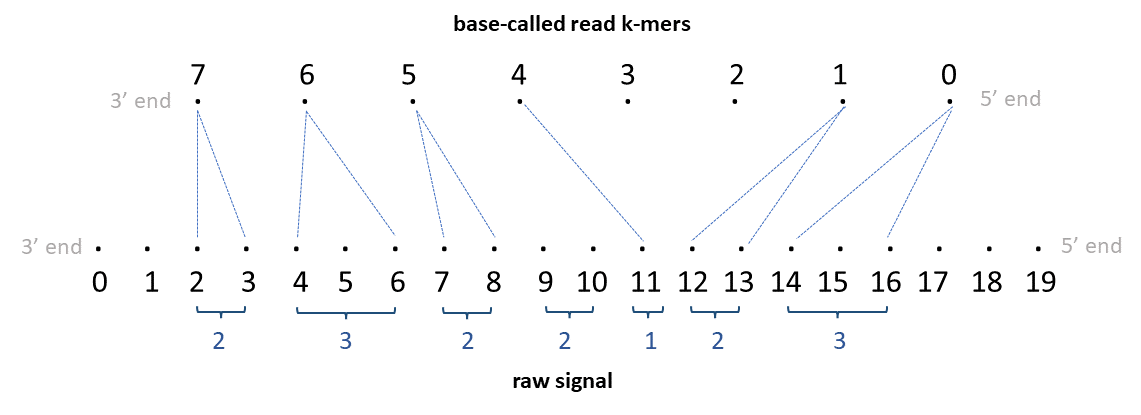
(Both examples are copied from f5c's documentation: https://hasindu2008.github.io/f5c/docs/output#resquiggle)
Note: To obtain the ss/si tags, one can add the -a option to f5c eventalign or the --bam-f5c to uncalled4 align.
One can use NanoporeSignals to create SignalAlignment objects from SAM/BAM reads. The alignment basically map signal intervals to sequence intervals (k-mers).
Some important things to bear in mind regarding the representation of the alignments when using the library:
- The
ss/sitags use 0-based indexing, while in Julia indexing is 1-based. - Intervals are closed (inclusive) on both sides.
- The sequence indices in the
ss/sirefer to the first position of the k-mer. NanoporeSignals generally keeps the full ranges of the k-mer. Operations such as slicing are based on interval overlaps, so they may return intervals that are only partially overlapping the query. - In most cases, a signal interval is mapped to a single k-mer. However, sequence insertions with more than one position will form a single sequence interval. E.g. in the DNA example shown above, the 0-based sequence insertion [4, 5] will be represented as the interval [5, 6+kmer_size-1] in NanoporeSignals.
API
Index
NanoporeSignals.SignalAlignmentNanoporeSignals.SignalAlignmentNanoporeSignals.aligned_signalNanoporeSignals.aligned_signalNanoporeSignals.aligned_signal_summaryNanoporeSignals.fast_alignmentNanoporeSignals.get_paNanoporeSignals.get_raw_readNanoporeSignals.isreverseNanoporeSignals.kmer_to_sigNanoporeSignals.polyA_rangeNanoporeSignals.pos_to_sigNanoporeSignals.readNanoporeSignals.scale_signal_f5cNanoporeSignals.segment_fftNanoporeSignals.segment_ttestNanoporeSignals.seq_endsNanoporeSignals.sequenceNanoporeSignals.sig_endsNanoporeSignals.sig_to_kmerNanoporeSignals.sig_to_posNanoporeSignals.sliceNanoporeSignals.steps
SignalAlignment
NanoporeSignals.SignalAlignment — Type
Nanopore signal to sequence alignement.
Fields:
read::XAM.XAMRecord: Reference to the read.sig_index::Union{SignalIndex, Missing}: Signal interval to k-mer interval index.seq_index::Union{SequenceIndex, Missing}: K-mer interval to signal interval index.reverse::Bool: True if the molecule is sequenced from 3′ to 5′ end (i.e. in dRNA-seq).kmer_size::Int: Size of the pore k-merkmer_center::Int: Which position to use as a representative for a k-mer
Implements: read, sequence, isreverse, seq_ends, sig_ends, steps
NanoporeSignals.SignalAlignment — Method
SignalAlignment(
read::XAM.XAMRecord;
kmer_size=missing,
kmer_center=missing,
pore=missing,
index=:both
)Get a SignalAlignment object for the given read. The function will try to parse the tags in the BAM record in order to find alignment data produced by a resquiggler.
Currently supported resquiggling formats:
- F5c style "si"/"ss" tags. These can be produced by F5c or Uncalled4.
The produced alignment maps k-mer intervals (a [start, end] pair of sequence positions) to raw signal intervals. The kmer_size should match the correct number for the pore used for sequencing. Since the library also provides for convenience functions that use positions instead of k-mer intervals, the kmer_center parameter is used for controlling which position of the k-mer is considered the "representative one". The parameters kmer_size and kmer_center can be set manually, or alternatively the pore parameter can be used to get preset values.
Arguments
read::XAM.XAMRecord: a XAM.XAMRecord with the read for which to get the signal alignment.kmer_size: How many neighbouring nucleotides influence a given measurement.kmer_center: Which position of the k-mer exerts the largest influence. This will be used to convert k-mer ranges to single positions.pore: Can be used instead of settingkmer_sizeandkmer_centermanually. In this case, preset values forkmer_sizeandkmer_centerwill be used, sourced from https://doi.org/10.1038/s41592-025-02631-4. Supported values::r9_4, :r10_4, :RNA001, :RNA002, RNA004.index: :sequence, :signal, or :both (default: :both) Used to control what indices to construct for accessing the alignment. Using only one index can limit the available functionality or degrade performance, but may be useful to reduce memory footprint.
NanoporeSignals.read — Method
read(sig_aln::SignalAlignment)Return the SAM/BAM record for the read.
Arguments
sig_aln::SignalAlignment: signal alignment.
NanoporeSignals.sequence — Method
sequence(sig_aln::SignalAlignment)Returns the nucleotide sequence of the read.
Arguments
sig_aln::SignalAlignment: signal alignment.
NanoporeSignals.isreverse — Method
isreverse(sig_aln::SignalAlignment)Returns true if the molecule is sequenced from 3′ to 5′ end (i.e. in dRNA-seq).
Arguments
sig_aln::SignalAlignment: signal alignment.
NanoporeSignals.seq_ends — Method
seq_ends(sig_aln::SignalAlignment)Returns the (first, last) sequence positions in the alignment.
Arguments
sig_aln::SignalAlignment: signal alignment.
NanoporeSignals.sig_ends — Method
sig_ends(sig_aln::SignalAlignment)Returns the (first, last) signal positions in the alignment.
Arguments
sig_aln::SignalAlignment: signal alignment.
NanoporeSignals.steps — Method
steps(sig_aln::SignalAlignment)Returns a vector of ((sig_start, sig_end), (seq_start, seq_end)) tuples representing the signal-to-sequence alignment with gaps in both signal and sequence.
Arguments
sig_aln::SignalAlignment: signal alignment.
fast_alignment
NanoporeSignals.fast_alignment — Method
fast_alignment(read::XAM.XAMRecord)Parse the SAM/BAM tags to get a minimalistic alignment in a fast manner.
Arguments
read::XAM.XAMRecord: SAM/BAM read with si/ss tags.
Returns
Vector with ((sig_start, sig_end), (seq_start, seq_end)) pairs. Gaps in the signal or sequence will be represented with missing.
Example
julia> fast_alignment(read)
127-element Vector{Tuple{Union{Missing, Tuple{Integer, Integer}}, Union{Missing, Tuple{Integer, Integer}}}}:
((10618, 10622), (424, 424))
((10623, 10697), (423, 423))
((10698, 10715), (422, 422))
((10716, 10737), (421, 421))
((10738, 10755), (420, 420))
⋮
((15575, 15618), (19, 19))
((15619, 15659), (18, 18))
((15660, 15666), (17, 17))
((15667, 15676), (16, 16))
((15677, 15696), (15, 15))slice
NanoporeSignals.slice — Method
slice(
sig_aln::SignalAlignment;
from::Integer=1,
to::Integer=typemax(Int64),
by::Symbol=:sequence
)Slice the SignalAlignment to a part of the alignment. Slicing can be done by :sequence or :signal:
:sequence- obtain a part of the alignment within a specific interval of sequence positions.:signal- obtain a part of the alignment within a specific interval of the raw signal.
Note: All k-mer intervals or signals overlapping the given interval will be included. E.g. an interval of 1 base pair can return 9 k-mers that include that position in RNA004, where the kmer_size is 9.
Arguments
sig_aln::SignalAlignment: signal alignment.from::Integer: start index of the slice (inclusive).to::Integer: end index of the slice (inclusive).by::Symbol: slice by :sequence or :signal.
kmer_to_sig
NanoporeSignals.kmer_to_sig — Method
kmer_to_sig(
sig_aln::SignalAlignment;
from::Integer=1,
to::Integer=typemax(Int64),
by::Symbol=:sequence
)Returns a generator for iterating over sequence k-mers to signal interval pairs. Gaps in the sequence will be skipped. The function allows limiting the results to specific sequence position or signal intervals. All intervals used for limiting the results are closed (inclusive) and any overlapping k-mer or signal interval will be included. Results will be sorted in increasing order of sequence positions.
Arguments
sig_aln::SignalAlignment: signal alignment.from::Integer: start index of the slice (inclusive).to::Integer: end index of the slice (inclusive).by::Symbol: slice by :sequence or :signal.
Examples
These are based on the examples from F5c's documentation:
- DNA: https://hasindu2008.github.io/f5c/docs/output#dna-example
- dRNA: https://hasindu2008.github.io/f5c/docs/output#direct-rna-example
(also shown in the Introduction above) Note: f5c uses 0-based indices and shows the k-mer start. We use 1-based indices and use the full k-mer interval. For this example, let's assume kmer_size=6 and kmer_center=3 (the third base of the 6-mer influences the signal the most).
DNA examples:
julia> kmer_to_sig(dna_aln; from=2, to=4) |> collect
4-element Vector{Pair{Tuple{Int64, Int64}, Tuple{Int64, Int64}}}:
(1, 6) => (3, 4)
(2, 7) => (5, 7)
(3, 8) => (8, 9)
(4, 9) => (12, 12)
julia> kmer_to_sig(dna_aln; from=2, to=5, by=:signal) |> collect
2-element Vector{Pair{Tuple{Int64, Int64}, Tuple{Int64, Int64}}}:
(1, 6) => (3, 4)
(2, 7) => (5, 7)dRNA examples:
julia> kmer_to_sig(rna_aln; from=2, to=4) |> collect
3-element Vector{Pair{Tuple{Int64, Int64}}}:
(1, 6) => (15, 17)
(2, 7) => (13, 14)
(3, 9) => missing
julia> kmer_to_sig(rna_aln; from=2, to=5, by=:signal) |> collect
2-element Vector{Pair{Tuple{Int64, Int64}, Tuple{Int64, Int64}}}:
(7, 12) => (5, 7)
(8, 13) => (3, 4)See also pos_to_sig for position-to-signal pairs and steps for full alignment (with sequence and signal gaps).
pos_to_sig
NanoporeSignals.pos_to_sig — Method
pos_to_sig(
sig_aln::SignalAlignment;
from::Integer=1,
to::Integer=typemax(Int64),
by::Symbol=:sequence
)Returns a generator for iterating over sequence position to signal interval pairs. K-mers are converted to a single position using the kmer_center parameter (e.g. k-mer (41, 49) will map to position 45 if kmer_center is 5). Gaps in the sequence will be skipped. The function allows limiting the results to specific sequence position or signal intervals. All intervals used for limiting the results are closed (inclusive) and any overlapping position or signal interval will be included. Results will be sorted in increasing order of sequence positions.
Arguments
sig_aln::SignalAlignment: signal alignment.from::Integer: start index of the slice (inclusive).to::Integer: end index of the slice (inclusive).by::Symbol: slice by :sequence or :signal.
Examples
These are based on the examples from F5c's documentation:
- DNA: https://hasindu2008.github.io/f5c/docs/output#dna-example
- dRNA: https://hasindu2008.github.io/f5c/docs/output#direct-rna-example
(also shown in the Introduction above) Note: f5c uses 0-based indices and shows the k-mer start. We use 1-based indices and use the full k-mer interval. For this example, let's assume kmer_size=6 and kmer_center=3 (the third base of the 6-mer influences the signal the most).
DNA examples:
julia> pos_to_sig(dna_aln; from=2, to=4) |> collect
2-element Vector{Pair{Int64, Tuple{Int64, Int64}}}:
3 => (3, 4)
4 => (5, 7)
julia> pos_to_sig(dna_aln; from=2, to=5, by=:signal) |> collect
2-element Vector{Pair{Int64, Tuple{Int64, Int64}}}:
3 => (3, 4)
4 => (5, 7)dRNA examples:
julia> pos_to_sig(rna_aln; from=2, to=4) |> collect
2-element Vector{Pair{Int64, Tuple{Int64, Int64}}}:
3 => (15, 17)
4 => (13, 14)
julia> pos_to_sig(rna_aln; from=2, to=5, by=:signal) |> collect
2-element Vector{Pair{Int64, Tuple{Int64, Int64}}}:
9 => (5, 7)
10 => (3, 4)See also kmer_to_sig for kmer-to-sequence pairs and steps for full alignment (with sequence and signal gaps).
sig_to_kmer
NanoporeSignals.sig_to_kmer — Method
sig_to_kmer(
sig_aln::SignalAlignment;
from::Integer=1,
to::Integer=typemax(Int64),
by::Symbol=:signal
)Returns a generator for iterating over signal interval to k-mer pairs. Gaps in the signal will be skipped. The function allows limiting the results to specific signal or sequence position intervals. All intervals used for limiting the results are closed (inclusive) and any overlapping k-mer or signal interval will be included. Results will be sorted in increasing order of signal intervals.
Arguments
sig_aln::SignalAlignment: signal alignment.from::Integer: start index of the slice (inclusive)to::Integer: end index of the slice (inclusive).by::Symbol: slice by :sequence or :signal.
Examples
These are based on the examples from F5c's documentation:
- DNA: https://hasindu2008.github.io/f5c/docs/output#dna-example
- dRNA: https://hasindu2008.github.io/f5c/docs/output#direct-rna-example
(also shown in the Introduction above) Note: f5c uses 0-based indices and shows the k-mer start. We use 1-based indices and use the full k-mer interval. For this example, let's assume kmer_size=6 and kmer_center=3 (the third base of the 6-mer influences the signal the most).
DNA example
julia> sig_to_kmer(dna_aln; from=2, to=12) |> collect
5-element Vector{Pair{Tuple{Int64, Int64}}}:
(3, 4) => (1, 6)
(5, 7) => (2, 7)
(8, 9) => (3, 8)
(10, 11) => missing
(12, 12) => (4, 9)
julia> sig_to_kmer(dna_aln; from=2, to=5, by=:sequence) |> collect
5-element Vector{Pair{Tuple{Int64, Int64}}}:
(3, 4) => (1, 6)
(5, 7) => (2, 7)
(8, 9) => (3, 8)
(10, 11) => missing
(12, 12) => (4, 9)dRNA example
julia> sig_to_kmer(rna_aln; from=2, to=12) |> collect
5-element Vector{Pair{Tuple{Int64, Int64}}}:
(3, 4) => (8, 13)
(5, 7) => (7, 12)
(8, 9) => (6, 11)
(10, 11) => missing
(12, 12) => (5, 10)
julia> sig_to_kmer(rna_aln; from=2, to=5, by=:sequence) |> collect
3-element Vector{Pair{Tuple{Int64, Int64}, Tuple{Int64, Int64}}}:
(12, 12) => (5, 10)
(13, 14) => (2, 7)
(15, 17) => (1, 6)See also sig_to_pos for signal-to-pos pairs and steps for full alignment (with sequence and signal gaps).
sig_to_pos
NanoporeSignals.sig_to_pos — Method
sig_to_pos(
sig_aln::SignalAlignment;
from::Integer=1,
to::Integer=typemax(Int64),
by::Symbol=:signal
)Returns a generator for iterating over signal interval to sequence position pairs. K-mers are converted to a single position using the kmer_center parameter (e.g. k-mer (41, 49) will map to position 45 if kmer_center is 5). Gaps in the signal will be skipped. The function allows limiting the results to specific signal or sequence position intervals. All intervals used for limiting the results are closed (inclusive) and any overlapping position or signal interval will be included. Results will be sorted in increasing order of signal intervals.
Arguments
sig_aln::SignalAlignment: signal alignment.from::Integer: start index of the slice (inclusive)to::Integer: end index of the slice (inclusive).by::Symbol: slice by :sequence or :signal.
Examples
These are based on the examples from F5c's documentation:
- DNA: https://hasindu2008.github.io/f5c/docs/output#dna-example
- dRNA: https://hasindu2008.github.io/f5c/docs/output#direct-rna-example
(also shown in the Introduction above) Note: f5c uses 0-based indices and shows the k-mer start. We use 1-based indices and use the full k-mer interval. For this example, let's assume kmer_size=6 and kmer_center=3 (the third base of the 6-mer influences the signal the most).
DNA example
julia> sig_to_pos(dna_aln; from=2, to=12) |> collect
5-element Vector{Pair{Tuple{Int64, Int64}}}:
(3, 4) => 3
(5, 7) => 4
(8, 9) => 5
(10, 11) => missing
(12, 12) => 6
julia> sig_to_pos(dna_aln; from=2, to=12, by=:sequence) |> collect
7-element Vector{Pair{Tuple{Int64, Int64}}}:
(3, 4) => 3
(5, 7) => 4
(8, 9) => 5
(10, 11) => missing
(12, 12) => 6
(13, 14) => 9
(15, 17) => 10dRNA example
julia> sig_to_pos(rna_aln; from=2, to=12) |> collect
5-element Vector{Pair{Tuple{Int64, Int64}}}:
(3, 4) => 10
(5, 7) => 9
(8, 9) => 8
(10, 11) => missing
(12, 12) => 7
julia> sig_to_pos(rna_aln; from=2, to=12, by=:sequence) |> collect
7-element Vector{Pair{Tuple{Int64, Int64}}}:
(3, 4) => 10
(5, 7) => 9
(8, 9) => 8
(10, 11) => missing
(12, 12) => 7
(13, 14) => 4
(15, 17) => 3See also sig_to_kmer for signal-to-kmer pairs and steps for full alignment (with sequence and signal gaps).
aligned_signal
NanoporeSignals.aligned_signal — Method
aligned_signal(
sig_aln::SignalAlignment,
slow5::Slow5.Slow5File;
to_pa::Bool=false,
read_id::Union{String, Missing}=missing,
read_id_tag::String="pi"
)Returns the raw signal segments, aligned to the sequence positions, e.g.
[
(1, [899, 886, 887, 903, 890]),
(2, [1210, 1199, 1213, 1202])
]Arguments
sig_aln::SignalAlignment: SignalAlignment instance.slow5::Slow5.Slow5File: A handle to the SLOW5/BLOW5 file with the raw signal.to_pa::Bool: If true, will convert the signal to picoampers. Default: false.read_id::Union{String, Missing}: The read id in the SLOW5/BLOW5 file. If not provided, it'll try to extract it from the SAM/BAM using theread_id_tagparameter to query the SAM/BAM tags.read_id_tag::String: SAM/BAM tag with the id of the raw record in the SLOW5/BLOW5 file. A single raw signal sometimes may contain multiple molecules, which are going to be split during base calling. Dorado uses thepitag to store the parent read. Default: "pi".
NanoporeSignals.aligned_signal — Method
aligned_signal(
sig_aln::SignalAlignment,
slow5::Slow5.Read;
to_pa::Bool=false
)Returns the raw signal segments, aligned to the sequence positions, e.g.
[
(1, [899, 886, 887, 903, 890]),
(2, [1210, 1199, 1213, 1202])
]Arguments
sig_aln::SignalAlignment: SignalAlignment instance.slow5_record::Slow5.Read: A record from the SLOW5/BLOW5 with the raw data for the read.to_pa::Bool: If true, will convert the signal to picoampers. Default: false.
aligned_signal_summary
NanoporeSignals.aligned_signal_summary — Method
aligned_signal_summary(
sig_aln::SignalAlignment,
slow5::Slow5.Slow5File;
summary_functions=[mean],
to_pa=false,
read_id::Union{String, Missing}=missing,
read_id_tag::String="pi"
)Executes summary functions on the segments of the signal aligned to each position and returns a DataFrame with the results. The DataFrame will have the following columns:
- pos: sequence position.
- kmer_start: first position of the k-mer, aligned to the segment.
- kmer_end: last position of the k-mer, aligned to the segment.
- dwell: duration that the k-mer spent in the pore, in seconds.
- <summaries>: one column per given summary function. By default it will be only the mean level of the event.
Note: The data frame will be according to the sequencing direction of the signal and the position and kmer columns will have missing values for sequence deletions (regions of the signal that didn't align to any sequence).
Arguments
sig_aln::SignalAlignment: SignalAlignment instance.slow5::Slow5.Slow5File: A handle to the SLOW5/BLOW5 file with the raw signal.summary_functions: Functions that will be evaluated on each event in the signal. Can be a vector of functions or :name => fn pairs.to_pa::Bool: If true, will convert the signal to picoampers. Default: false.read_id::Union{String, Missing}: The read id in the SLOW5/BLOW5 file. If not provided, it'll try to extract it from the SAM/BAM using theread_id_tagparameter to query the SAM/BAM tags.read_id_tag::String: SAM/BAM tag with the id of the raw record in the SLOW5/BLOW5 file. A single raw signal sometimes may contain multiple molecules, which are going to be split during base calling. Dorado uses thepitag to store the parent read. Default: "pi".
Examples
E.g. for dRNA sequencing which goes in 3′->5′ direction:
julia> aligned_signal_summary(
aln,
blow5;
read_id=BAM.tempname(reads[1]),
to_pa=true,
summary_functions=[mean, :geometric_mean => xs -> reduce(*, xs)^(1/length(xs))])
125×6 DataFrame
Row │ pos kmer_start kmer_end dwell mean geometric_mean
│ Int64? Int64? Int64? Float64 Float64 Float64
─────┼─────────────────────────────────────────────────────────────────
1 │ 429 424 432 0.00125 69.2437 69.1985
2 │ 428 423 431 0.01875 91.5471 91.2353
3 │ 427 422 430 0.0045 102.751 102.495
4 │ 426 421 429 0.0055 90.1499 89.9085
5 │ 425 420 428 0.0045 68.6686 68.3237
⋮ │ ⋮ ⋮ ⋮ ⋮ ⋮ ⋮
121 │ 24 19 27 0.011 67.7238 67.3204
122 │ missing missing missing 0.01025 58.6041 58.4725
123 │ 22 17 25 0.00175 63.3077 63.3012
124 │ 21 16 24 0.0025 67.4599 67.3375
125 │ 20 15 23 0.005 90.1732 89.9791get_pa
NanoporeSignals.get_pa — Method
get_pa(blow5_record::Slow5.Read)Returns the read's signal converted to picoampers.
pA = (raw_signal + offset) * range / digitisation))
Arguments
blow5_record::Slow5.Read: a SLOW5/BLOW5 record.
scale_signal_f5c
NanoporeSignals.scale_signal_f5c — Method
scale_signal_f5c(pa_signal::Vector{Float64}, read::XAM.XAMRecord)Get the signal scaled to the model using scaling coefficients from f5c. Expects a signal in picoampers and the BAM record for the read populated with f5c tags. The formula is:
scaled pA current values = (pA - sh) / sc
where pA is the signal in picoampers and sh and sc are f5c scaling parameters written as tags to the SAM/BAM.
Arguments
pa_signal::Vector{Float64}: signal in picoampers.read::XAM.XAMRecord: SAM/BAM record that has f5c tags (shandsc).
get_raw_read
NanoporeSignals.get_raw_read — Method
get_raw_read(
read::XAM.XAMRecord,
slow5::Slow5.Slow5File;
read_id::Union{String, Missing}=missing,
read_id_tag::String="pi"
)Returns the SLOW5/BLOW5 record associated with the given SAM/BAM read.
If read_id is provided it'll look for record with that id. Otherwise, it'll try to find the SLOW5/BLOW5 record id by looking at the read_id_tag tag in the SAM/BAM record. If the read_id_tag is missing from the record, it'll try to look for a SLOW5/BLOW5 record with id equal to the SAM/BAM QNAME.
Arguments
read::XAM.XAMRecord: SAM/BAM record for which to find the SLOW5/BLOW5 record.slow5::Slow5.Slow5File: A handle to the SLOW5/BLOW5 file with the raw signal.read_id::Union{String, Missing}: The read id in the SLOW5/BLOW5 file. If not provided, it'll try to extract it from the SAM/BAM using theread_id_tagparameter to query the SAM/BAM tags.read_id_tag::String: SAM/BAM tag with the id of the raw record in the Slow5/Blow5 file. A single raw signal sometimes may contain multiple molecules, which are going to be split during base calling. Dorado uses thepitag to store the parent read. Default: "pi".
segment_ttest
NanoporeSignals.segment_ttest — Method
segment_ttest(
signal::Vector{<:Real};
window::Integer=18,
pvalue_threshold::AbstractFloat=0.05,
clip::Integer=2
)A simple signal segmentation strategy that slides two windows of the given size over the signal and performs a t-test between them at each position to find breaking points. The function will then run peak calling to find the positions of the most significant p-values (discarding the ones above pvalue_threshold).
Arguments
signal::Vector{<:Real}: A vector with the signal measurements.window: Size of the window. Two adjacent windows of the given size will be used.pvalue_threshold: Less significant positions will be ignored from the peak calling. I.e. positions with a t-test p-value >pvalue_threshold.clip: Clip the values in each window to ignore likely "transition values".
segment_fft
NanoporeSignals.segment_fft — Method
segment_fft(
signal::Vector{<:Real},
sampling_rate;
cutoff::Integer=180,
window=10,
threshold=5,
clip=2
)A simple signal segmentation strategy that denoises the signal using fast Fourier transform and then looks for breakpoints using two attached sliding windows and looking for large enough differences between them.
Warning: this function is experimental.
Arguments
signal::Vector{<:Real}: The raw signal in pA.cutoff::Integer: cutoff frequency.sampling_rate: sampling rate for the read (available in the SLOW5/BLOW5 record).window: Window size.threshold: Minimum signal difference between the two windows for detecting a breakpoint.clip: The number of measurements that will be clipped from the ends of the windows, as they are likely to be representative of the transition state.
polyA_range
NanoporeSignals.polyA_range — Method
polyA_range(
aln::SignalAlignment,
seq::String,
signal::Vector{<:Real};
min_len=200,
homoA_level_low=70,
homoA_level_high=100,
block=10
)A quick and simple heuristic algorithm for finding the start and end of the poly(A) region in a signal. The method essentially will start from the signal region aligned to the last non-poly(A) k-mers of the sequence and look back until it finds a region resembling a poly(A) stretch. It'll then expand in both directions until it reaches levels outside the expected bounds.
Warning: this function is experimental.
Arguments
aln::SignalAlignment: the signal alignment.seq::String: the sequence of the read.signal::Vector{<:Real}: the raw signal for the read in picoampers.min_len: minimum length of the poly(A) region (in sample points).homoA_level_low: Minimum level for the poly(A) signal intensity.homoA_level_high: Maximum level for the poly(A) signal intensity.block: Number of sample points that will be used to test if the signal deviates from the expected level.
Examples
julia> blow5 = slow5_open("raw_reads.blow5", "r")
julia> slow5_idx_load(blow5)
julia> read = first(open(BAM.Reader, "reads.bam"))
julia> aln = SignalAlignment(read; pore=:RNA004)
julia> raw_read = get_raw_read(read, blow5)
julia> signal = get_pa(raw_read)
julia> polyA_start, polyA_end = polyA_range(aln, string(BAM.sequence(read)), signal)
(3286, 4953)